These days I’m writing up the discussion of my sensitivity analysis paper on missing data using the Total Evidence method (more about it here and here). One evident opening for proposing future improvement on my analysis is the obvious “let’s-do-it-again-with-more-data” one… But a recent Science paper by Jarvis et al made me reconsider that. Is more the always better?
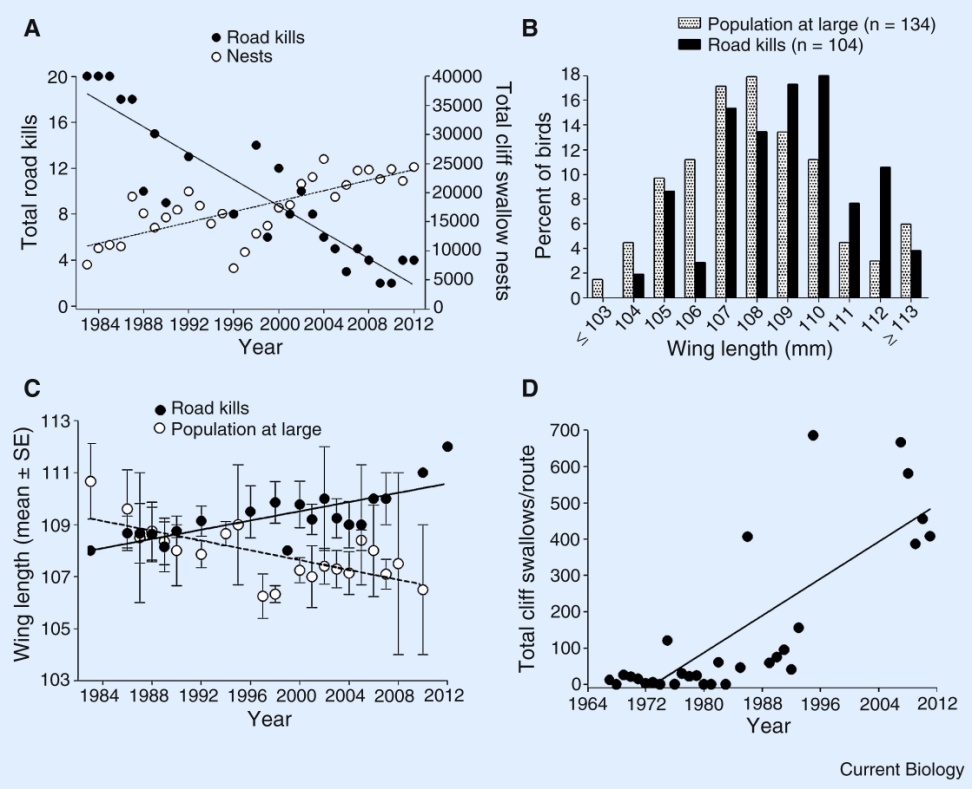
Jarvis and his numerous colleagues just published one of the biggest bird phylogenies that contrasts with the previous reference one (by Jetz et al in Nature). In Jetz’s paper, the authors were interested in the relations among modern birds (read “non-dinosaurs ones”) and tackled the question by trying to sample the whole of bird biodiversity (9,993 species!). However, as in most analyses of this kind, the molecular data can be fairly poor (note that they still managed to collect a maximum of 15 genes for 6663 species). Even though the global picture of avian diversity is clear, some regions are less resolved than others and an obvious way to fix that would be to sample more genes per species. And that is, in a way, exactly what Jarvis and his colleagues tried to achieve.
In this new study, the authors went on sampling not 15, 70 or 150 genes but 8251 genes per species! This led to a really deep and long analysis – over 400 CPU years, and I thought 150 was long! – of the complete genome of birds. By the way, they use the name Total Evidence nucleotide tree (TENT) to design the results of their analysis which is pretty confusing since a total evidence tree means something quite different to me. But that’s just a semantic rant. Using this massive TENT, the authors fixed some previously poorly resolved nodes, redefined the names of ancient divergences among birds (with the Passerea – tits and relatives – and the Columbea – pigeons and relatives), demonstrated an explosive (“big-bang”) radiation after the K-T event and determined the patterns of certain traits evolution (such as raptoriality or vocal learning). In short a thorough work that allowed the authors to say: “The conflict we observe with other data types can no longer be considered to be due to error from smaller amounts of sequence data”. I feel that writing something like that in a paper is a nice achievement!
However – don’t get me wrong, this paper is yet a great example of collaborative work and insight in new methods – the sample size is… 45 species. In other words, Jetz et. al sampled 100% of the species but less than 1% of the data as for Jarvis et al., they sampled 100% of the data for less than 1% of the species. In this case, we have two extreme views of the same question (“how did avian diversity evolve?”) and in both cases, I think the macroevolutionary claims are weakened by the number of species or the amount of data… However, from a practical point of view, I think the method that included more species will be preferred by researchers since their species of interest are more likely to be present in that tree. What’s the best balance? Full genome or full sampling? I’ll leave it to you to decide…
Author
Thomas Guillerme, guillert[at]
Photo credit
http://everythingbirdsonline.com/